
Mingzhou (Joe) Song
Department of Computer Science, New Mexico State University, P. O. Box 30001, MSC CS, Las Cruces, New Mexico 88003, U.S.A.
Zhengyu Ouyang
Department of Computer Science, New Mexico State University, P. O. Box 30001, MSC CS, Las Cruces, New Mexico 88003, U.S.A.
Z. Lewis Liu
National Center for Agricultural Utilization Research, U.S. Department of Agriculture, Agriculture Research Service, 1815 N University Street, Peoria, Illinois 61604, U.S.A.
Associated Data
Abstract
Composed of linear difference equations, a discrete dynamical system model was designed to reconstruct transcriptional regulations in gene regulatory networks for ethanologenic yeast Saccharomyces cerevisiae in response to 5-hydroxymethylfurfural, a bioethanol conversion inhibitor. The modeling aims at identification of a system of linear difference equations to represent temporal interactions among significantly expressed genes. Power-stability is imposed on a system model under the normal condition in the absence of the inhibitor. Non-uniform sampling, typical in a time course experimental design, is addressed by a log-time domain interpolation. A statistically significant discrete dynamical system model of the yeast gene regulatory network derived from time course gene expression measurements by exposure to 5-hydroxymethylfurfural, revealed several verified transcriptional regulation events. These events implicate Yap1 and Pdr3, transcription factors consistently known for their regulatory roles by other studies or postulated by independent sequence motif analysis, suggesting their involvement in yeast tolerance and detoxification of the inhibitor.
1 Introduction
Quantitative modeling of gene regulatory networks (GRNs) in ethanologenic yeast using high throughput biotechnology, in part a large-scale computational problem, holds the key towards high-yield ethanol production from biomass in the presence of inhibitory chemical compounds. By far, few data-driven approaches are capable of describing the information flow over time in a dynamical biological system. Dynamical system modeling of GRNs, however, empowers one to understand systematically the interactions among variables in a system. Motivated by its computational feasibility for modeling large-scale dynamical systems, we study the discrete dynamical system (DDS) model, composed of linear difference equations, for reconstruction of GRNs in yeast during biomass conversion to ethanol. The Verhulst equation, a single-variable DDS model, is an example that is widely used in mathematical biology [] to study population dynamics. Although DDS modeling has been utilized for GRN reconstruction by estimating system coefficients using least squares [], their potential has remained largely unrecognized in molecular systems biology. Only until recently, gene interactions or biochemical reaction pathways by DDS models consisting of either linear difference equations or finite state linear equations have been characterized [, , ]. Our work moves along with three innovations. The first is to perform log-time domain interpolation on non-uniformly spaced samples and resample from equally spaced time locations. The second is to assess statistical significance of all feasible linear difference equations for a given gene variable and to choose the most significant one, as well as to assess the statistical significance of the overall DDS model. The third is to enforce power stability on the DDS model so that it does not exhibit chaotic or unstable behaviors under a normal condition. A DDS is power stable if variables in the system stay bounded as time goes to infinity given any finite initial state.
Our work has originated from the investigation of genetic mechanisms for bioethanol conversions in yeast in pursuit of renewable sources of energy. As public interests in alternative sources of energy rise, agriculture as a renewable energy producer has soared. Biomass, including lignocellulosic materials and agricultural residues, has become a focus of low-cost materials for biofuel production. One major barrier of biomass conversion to ethanol is inhibitory compounds produced during biomass pretreatment, which interfere with microbial growth and subsequent fermentation. For economic reasons, dilute acid hydrolysis is commonly used to prepare the biomass degradation for enzymatic saccharification and fermentation [, ]. However, numerous side-products are generated by this pretreatment, many of which inhibit microbial metabolism. More than 100 compounds are known to have potential inhibitory effects on microbial fermentation []. Among these compounds, 5-hydroxymethylfurfural (HMF) and furfural are the most potent and representative inhibitors derived from biomass pretreatment [, ]. Few yeast strains tolerant of inhibitors are available. The molecular mechanisms involved in the stress tolerance and detoxification are not well understood for yeast. Based on transcriptome pofiling analysis, a concept of genomic adaptation to the biomass conversion inhibitors by the ethanologenic yeast has been proposed [, ]. However, a great deal of detailed knowledge about GRNs in yeast involved in stress tolerance during the biomass conversion still remains unknown.
In the computational and biological context described above, we have developed DDS models to study the genetic basis underlying metabolic pathway of the ethanologenic yeast. As initiated in this study, we have delineated through DDS models how yeast behaves in response to the inhibitor HMF during the earlier exposure to the inhibitor for ethanol production. In this model, the change rate in expression level of a target gene at a discrete time point is a linear function of the expression levels of influential genes at the previous discrete time point. This model facilitates the characterization of gene interactions in ethanol production by yeast under both control and HMF-stress-treatment conditions, allowing one to introduce specific perturbations into a system and predict the effects on biomass conversion under various stress conditions. Furthermore, the model provides potential to identify relevant genes and gene interactions for optimal genetic manipulations that will guide the engineering of more robust yeast strains for economic ethanol production.
DDS modeling is advantageous given the increased availability of experimental designs that collect time-course gene expressions at the whole-genome scale, though other modeling paradigms exist for different emphases:
-
Temporal probabilistic networks – The dynamic Bayesian network (DBN) is an extension of Bayesian networks, which incorporates time transitions between Bayesian networks. A DBN can describe statistical and temporal dependencies among genes in a GRN. We have no doubt that DBNs are successful in extracting probabilistic dependencies in modeling GRNs [, , ]. Although certain DBNs can be converted to probabilistic Boolean networks [], DBN modeling is an indirect tool to understand system dynamics since it does not explicitly describe temporal relations among genes in a functional form.
-
Continuous dynamical system models – Differential equations in both deterministic [, ] and stochastic [] formulations have been used to model interactions in GRNs in continuous time. The E-CELL Project [, , ] targets at knowledge-based reproduction, not data-driven reconstruction, of intracellular biochemical and molecular interactions within a single cell using differential equations. The stochastic master equations relate state probabilities by differential equations, impractical for biological systems involving many variables because of the computational burden. Recent research has been focusing on improving the scalability of such models []. However, almost all differential equations reduce to difference equations in practical applications. Direct DDS modeling overrides this intermediate step and speaks the native discrete time language of a computer. We believe it is more effective to go without the intermediate mode of differential equations. In addition, the actual time interval between discrete time points in difference equations can be adjusted to the sparsity of data, making it more flexible to model the dynamics at different resolutions.
-
Boolean networks – In a Boolean network [, , , , ] and its Markovian [] or probabilistic [] extensions, each variable takes the value of either 0 or 1. The dichotomous nature of a Boolean network seriously limits its capacity to discriminate quantitative differences. This can be crucial when such differences are more interesting than the mere information of presence (1) or absence (0). Our primary goal is to establish a system model encoding regulatory mechanisms in biomass conversion to ethanol, especially the quantitative shift of biotransformation and detoxification of the inhibitors for effective ethanol conversion, which requires information beyond the presence or absence of genes. Thus, Boolean networks are not the best dynamical strategy to describe accurately the amount of biotransformation as a function of dynamical metabolic interactions. This has been indicated by the recent extension of probabilistic Boolean networks to incorporate more than two levels for each variable [].
Following the introduction, we consider DDS modeling in Section 2 and present our solution to data-driven modeling that creates models of statistical significance, including a log-time domain interpolation to address the issue of non-uniformly sampled time points. The scaling performance of our DDS modeling is evaluated through a simulation study in Section 3. We discuss the reconstructed GRN in the context of known transcriptional regulations and suggest potentially novel gene interactions of yeast in response to HMF in Section 4. Finally, we give conclusions and potential future work in Section 5.
2 The discrete dynamical system model
Although dynamics in molecular processes are largely nonlinear as in various kinetics models, the number of observations sufficient to induce a nonlinear model for a biological system is too large to be practical for a system with more than a handful of variables. Instead of nonlinear models, we use a first-order linear DDS model to capture system dynamics. A system can be approximated using a linear DDS model when the perturbation to the system is sufficiently small. In our experiment, the time course gene expression we collected reflected the initial response of gene expressions to the inhibitor HMF before saturation, a major nonlinear dynamical effect, takes place. Thus, we consider the DDS model capable of approximating primary expression response to HMF.
In a first-order linear DDS model, the transition from one state at discrete time t – 1 to the next state at t depends linearly on the state of the system at only time t – 1. Let h be the constant time span of 1 unit of discrete time. First order refers to the transition from t – 1 to t does not depend on the state of the system at t – 2, t – 3, and so on, but the state at t – 1. Let g[t] = (g1[t], g2[t], …, gN[t])⊤ be a vector of the expression levels of N genes at time t. Let e[t] = (e1[t], e2[t], …, eK[t])⊤ be a vector of the strength of K external stimuli at time t. A first-order linear DDS model is defined by
where A = {aij} is an N × N system matrix and aij (i ≠ j) is the influence of gene j on gene i, aii is the self-control rate, B = {bik} is an N × K external influence matrix where bik is the influence of the k-th stimulus on gene i, ∈⃗ [t] = (∈1[t], ∈2[t], …, ∈N[t])⊤ is a vector of noise levels to each gene at time t. The noise is estimated by fitting the DDS model, and thus is a function of the time interval as well as the observed data. In the modeling process, we assume the noise model Gaussian. We also introduce a possible intercept vector c to the right hand side of the above equation during model selection for each node.
2.1 Estimating coefficients for each linear difference equation
From an experiment with trials under various external stimuli and in replica, one can collect M trials of time course observations or trajectories of a system at the discrete time points 0, 1, 2, …, T. Let gm[0], gm[1], …, gm[T] be the m-th observed trajectory of (m = 1 … M) the system, and em[0], em[1], …, em[T] be the m-th trajectory of external stimuli applied to the system. We use the least squares to find optimal estimates of system matrix A and external influence matrix B. The DDS model defined in can be written as a collection of all M trajectories by
where
Equivalently for each gene variable, we have the multiple linear regression form
where  i is a subset of indices to gene variables in the system, pointing to non-zero elements on row i of A, and
i is a subset of indices to gene variables in the system, pointing to non-zero elements on row i of A, and  i a subset of indices to external stimuli, pointing to nonzero elements on row i of B. Any coefficients not indexed in the subsets are considered zero. This is a critical explicit form in the DDS modeling in order to express only the most statistically significant subsets for influencing a gene.
i a subset of indices to external stimuli, pointing to nonzero elements on row i of B. Any coefficients not indexed in the subsets are considered zero. This is a critical explicit form in the DDS modeling in order to express only the most statistically significant subsets for influencing a gene.
Let ai = (ai1, …, aiN)⊤ and bi = (bi1, …, biK)⊤ be the parameters associated with gene variable i. ai and bi can be solved independently of other parameter vectors using the multiple regression in . By least squares, optimal estimates for ai and bi are
where ,
and
2.2 The most significant linear difference equation for each gene variable
In solving the multiple linear regression in for gene i, assigning {1, 2, …, N} to 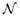 i and {1, 2, …, K} to
i and {1, 2, …, K} to  i will guarantee minimal least squares. However, such a solution by involving all variables as independent variables is in general not statistically significant due to the high degrees of freedom of the regression model. Thus obtained DDS model would most likely fit the dynamical behaviors caused by noise as well as those by consistent systematic interactions.
i will guarantee minimal least squares. However, such a solution by involving all variables as independent variables is in general not statistically significant due to the high degrees of freedom of the regression model. Thus obtained DDS model would most likely fit the dynamical behaviors caused by noise as well as those by consistent systematic interactions.
Hence we strategically reduce the number of variables involved in each difference equation so that the resulting fit can attain a given statistical significance. We achieve this selection by finding  i and
i and  i that together produce the smallest p-value of the F-test for each multiple linear regression. For N genes and K external stimuli, there are 2N+K −1 possible subsets– excluding the empty set as the null hypothesis–to consider for
i that together produce the smallest p-value of the F-test for each multiple linear regression. For N genes and K external stimuli, there are 2N+K −1 possible subsets– excluding the empty set as the null hypothesis–to consider for  i and
i and  i, only computationally feasible for a system with less than a dozen of variables. We limit the number of possible incoming edges or potential regulators for each variable to some computational doable number. Although this leads to an incomplete exploration of the system search space, our experience indicates that major influential gene variables can be identified even when the number of regulator nodes explored is small for typical sample sizes of a microarray experiment.
i, only computationally feasible for a system with less than a dozen of variables. We limit the number of possible incoming edges or potential regulators for each variable to some computational doable number. Although this leads to an incomplete exploration of the system search space, our experience indicates that major influential gene variables can be identified even when the number of regulator nodes explored is small for typical sample sizes of a microarray experiment.
Since the chance of making a Type I error increases dramatically as we increase the number of interactions to inspect, we perform multiple testing p-value adjustment by Bonferroni correction. The p-value for the multiple linear regression of each node is multiplied by the total number of regressions performed in the entire modeling process to derive the adjusted p-value. This p-value is capped to one if the product is greater. The p-value for each coefficient in a single difference equation is also inflated in exactly the same way. The Bonferroni adjustment provides the most stringent criterion among all alternatives and only the most highly significant interactions represented by multiple linear regressions can survive the p-value cutoff after inflation.
2.3 Stabilization
Although solutions to the linear difference equations constitute an optimal fit to the observed data, the resulting DDS model can be unstable, meaning that the log expression levels of some genes increase to infinity or decrease to negative infinity as time goes on when the initial state of the system is finite. Thus we stabilize the system model when no external stimuli are present.
Now we derive the stabilization formula. Equivalently, can be written as
When the system is not subject to external stimuli or noise, it becomes
In the bioethanol conversion process, this system equation describes the ideal behavior of yeast gene expression without the inhibitor HMF in a zero-noise environment. In such a system, one does not expect the expression of any gene becomes unstable during the experiment since otherwise the subject perishes. An optimal solution found for A by may lead to an unstable system in . Let W = hA + I. A necessary and sufficient condition for the system described by to be stable is to require W to be power stable – all eigenvalues of W must be located within or on the unit circle; or the spectral norm must be no greater than one. Let λ(W) be the sequence of eigenvalues of W. The spectral norm ρ(W) is defined by []
Let Λ be a diagonal matrix, generated by diag(λ(W)), and V be a matrix whose columns are the eigenvectors in an order corresponding to the order of eigenvalues in λ(W). It follows that
We stabilize W to obtain Ws by scaling all its eigenvalues by its spectral norm if the spectral norm is greater than 1, while maintaining the same eigenvectors, that is,
Let As be the transformed matrix A after stabilization. Plugging in the definition of W, we obtain
if the spectral norm of W is greater than 1. Replacing A by As in , we obtain
There are several theoretical and numerical properties associated with our stabilization strategy. It is evident that any coefficients off the diagonal line in A with a value close to 0 will be closer to 0 after stabilization. This ensures that no new interactions between different genes will be introduced by stabilization. The spectral norm can be found efficiently using the power method without obtaining all eigenvalues or eigenvectors of matrix W. In addition, since there is no matrix decomposition involved, the stabilized matrix As will be real if A is real, which holds true theoretically but could be violated numerically by other approaches.
2.4 Statistical significance of a discrete dynamical system model
Let the minimum p-value of fitting a linear difference equation to gene i be pi. The p-value of a fitted DDS model is computed by
where pi is computed by the F-tests during the fitting of linear model for gene variable i. This defines a conservative p-value. Nevertheless, the p-value of a DDS model is a statistically effective and computationally efficient measure to determine the chance an estimated model would arise randomly. This p-value is influenced by 1) how well each linear difference equation can be fitted to the data and 2) the number of nonzero coefficients in the model, which constitute two competing factors. Our algorithm minimizes the p-value by trade-off between both factors.
2.5 Log-time interpolation
Non-uniform time sampling is often used in a time course experimental design, such that various frequency components in the original continuous signal can be preserved adaptively. Conversely, interpolation in the original time domain over non-uniform samples tends to distort high frequency components in the original signal. To save sharp transitions at densely sampled time locations, we apply a logarithm transform on time by
where t′ is the time variable in the log-time domain. Selection of the constant t0 is determined by how well it equalizes the distance between each consecutive pair of time points after the log-time transform. The observed samples are then interpolated by cubic splines in the log-time domain, by assuming that the sampling times are designed sufficiently well to capture major change of the stimuli; or equivalently, the change of gene expression levels between two consecutive time points can be captured by the cubic splines. Let x = f(t′) be the interpolated cubic spline. One can obtain values at equally spaced time points 0, h, 2h, …, qh, …, in the original time domain by
where h is the sampling interval. We pick the same number of interpolated points as the number of points in the original data set. So the interpolation solely serves to equalize the non-uniform time points in the log-time domain. If more points were interpolated, the p-value must be adjusted to that effect, otherwise, faulty significance might arise. The DDS model will be fitted to the interpolated values in the original time domain, using the procedure described in previous subsections.
3 Simulation study of the scaling performance of DDS modeling
The scaling property of DDS modeling determines its applicability to a wide range of systems. Through a simulation study, we demonstrate the scaling performance of our DDS reconstruction method under different network sizes, or numbers of variables. The measures we use to evaluate the performance include the false negative rate (FNR) and the false discovery rate (FDR), and the Hamming distance. We define an interaction as an ordering from node j to node i such that entry aij at the i-th row and the j-th column in system matrix A is not zero. The FNR is the ratio of the total number of missed interactions to the total number of original interactions. The FDR is the ratio of the number of incorrectly detected interactions to the number of detected interactions. We do not include the false positive rate (FPR) here because it is usually magnitude lower than FDR when a system is sparsely connected as in many biological systems. The FNR and FDR qualitatively evaluate whether the topology of a DDS model has been correctly identified. The Hamming distance is defined as the total number of false negative and false positive edges in the reconstructed network in reference to the original ground truth network. Hamming distance can be interpreted as a measure to determine how two graphs mismatch each other topologically – the greater the distance, the severer the mismatch.
For each given network size N, a random N × N system matrix A can be generated with the following specifications. For each row, 2 or 3 entries are selected randomly and uniformly from {1, …, N}. The values in each of the selected entries are also determined randomly and uniformly from [−10, 10]. All remaining entries in the row are set to zero. Then the system is stabilized, as described in Section 2.3, by scaling all eigenvalues of matrix hA + I, where I is an N × N identity matrix, to be on or within the unit circle. No scaling is done if all eigenvalues are already on or within the unit circle.
In simulation of a DDS model, we consider two types of noises: the random biological variability ∈b and the random measurement error ∈m. We consider the final observed variable a sum of an original unobserved variable plus both noises. The biological noise influences the system dynamic, while the measurement noise does not. Let g[t] be a state vector for observed values of all nodes at time t, containing both noises. Let gb[t] be the unobserved state vector for all nodes at t, containing only biological noise. It is important to note that only gb[t] participates in the dynamical evolution of the system. Thus, we use the following DDS model characterized by A for gb[t]:
where ∈⃗b[t] is a random vector representing the biological noise distributed as at time t, which arises from the random biological variability. It is unnecessary to include external influence matrix B because it will not influence the scaling performance evaluation. Thereafter, a final trajectory can be obtained by adding the measurement noise to the biological state vector gb[t]
where random vector ∈⃗m[t], with each entry a random variable distributed as , represents the measurement error introduced by imprecision in instrumentation at time t.
To quantify the strength of noises, we define the signal to measurement noise ratio (SMNR) as 10 times log10 of the sum of squares of the signal divided by the sum of squares of the measurement noise. Analogically, we define the signal to biological noise ratio (SBNR). The units of both ratios are decibels (dB).
We studied the scaling performance using network sizes 32, 64, 128, 256, 512, and 1,024. For each network size, we generated five random DDS models to obtain an average performance. For each randomly generated DDS system, we simulated 4 trajectories, with 6 time points (h = 1) and the state vector randomly initialized at time zero, under a high noise setting (SBNR ≈ 10 dB, SMNR ≈ 10 dB) and a low noise setting (SBNR ≈ 20 dB, SMNR ≈ 20 dB). The choices of the network sizes, the sample size, and the trajectory length align with our experimental design of gene expression in yeast in response to HMF. Then we performed DDS modeling to reconstruct a system for each set of trajectories corresponding to an original DDS system. Figure 1 shows the average scaling performance FNR and FDR with their standard errors under low and high noise settings. The monotonic FNR and FDR curves suggest that the scaling performance of DDS modeling in determining the correct topology decreases as the network size increases. In the high noise setting in Fig. 1(a), the FNR can range from 12% to 45%, and the FDR from 20% to 55%, as the network size increases from 32 to 1,024. In the low noise setting in Fig. 1(b), the FNR is less than 15% when the network size is 1,024; the FDR is about 20% when the network size is 128; both FDR and FNR reduce to around 5% when the network size is 32. If high noise prevails in an experiment, the strategy to improve the performance is to increase either the sample size or the number of time points during the transient phase of the underlying dynamical system.
The scaling performance, FNR and FDR, of DDS modeling as a function of the network size, under two different noise settings.
Table 1 shows the average Hamming distance as a function of the network size under the low and high noise settings. Each shown distance is an average from five instances of networks with the same size. Although the shown average Hamming distance increases almost linearly with the network size, the Hamming distance, if normalized by N2, would drop linearly as the network size increases, indicating the strength of our DDS modeling method.
Table 1
Average Hamming distance.
| Average Hamming Distance | ||
|---|---|---|
| Network Size | SBNR≈SMNR≈10dB | SBNR≈SMNR≈20dB |
| 32 | 28.2 | 8.6 |
| 64 | 93.6 | 29.6 |
| 128 | 230.8 | 100.2 |
| 256 | 544.4 | 249.2 |
| 512 | 1,333.0 | 534.0 |
4 Reconstructed gene regulatory networks of yeast in response to HMF
We performed DDS modeling on time-course microarray measurements of relative mRNA levels for transcriptional interactions among genes in yeast during the earlier exposure to the inhibitor HMF for ethanol production. After the initial exposure to HMF for about 2 hours, the expression profile of involved genes in yeast evolves to a saturation stage when linear gene interactions come to an end and strong nonlinear gene interactions dominate. Our objective is to detect earlier linear interactions using the DDS model, when gene expression changes are steady and not saturated. For complete details of the experiment design, microarray data analysis, gene clustering, and modeling results, please refer to the .
Experimental design
Target genome microarray of Saccharomyces cerevisiae was fabricated with a recent version of 70-mer oligo set representing 6,388 genes. Each genome microarray was designed with two replications on one microarray slide. Each microarray slide consisted of 13,000 elements including replicated target genes and spiking-in quality controls for linear dynamic calibration, ratio reference, DNA sequence background, and slide background controls. The first developed universal external RNA control was applied in microarray experiments []. Ethanologenic yeast S. cerevisiae NRRL Y-12632 was used and HMF added 6 hours after incubation []. A set of gene expression profiles derived from a yeast culture grown under the same conditions without the HMF treatment served as a control. The time point at inhibitor addition was designated as hour 0. Yeast cells were harvested at 0 hour, 10 min, 30 min, 1 hour, and 2 hour. The non-uniform sampling occurs densely at the beginning phase to allow one to capture accurately the initial dynamical response at the onset of external stimuli.
Microarray data analysis
Each microarray slide was scanned and data acquisition obtained using GenePix 4000B scanner and GenePix Pro software after normalization using universal RNA control []. Median of foreground signal intensity subtracted by background for each dye channel was used. Data were collected with two biological replications each with two technical replications. Based on ANOVA and a cluster analysis, 364 significantly differentially expressed genes by the HMF treatment were selected. We shifted the log-transformed microarray data on each chip by the median of the chip to correct system biases.
Gene clustering
Genes that have highly linearly correlated expression time courses can confuse DDS modeling. If these genes are treated as different variables, DDS modeling would pick only a single one while ignoring all others, leading to very different conclusions for how these genes influence others, though they are equivalent due to linearly correlated dynamical behaviors. Thus these genes should be treated as a single variable in DDS modeling. A representative from a cluster of linearly correlated genes can be designated as this single variable. In addition, selecting only one representative gene that resembles other genes the most from each cluster of linearly correlated genes will greatly reduce the computation in DDS reconstruction. We performed a clustering procedure from a package developed in the R language []. A total of 169 gene clusters and representative gene for each cluster were identified and are shown in Table 2.
Table 2
The 169 clusters of 364 genes. The first gene in each cluster is the representative.
| Cluster | Genes |
|---|---|
| C1 | ZRT2 SAM3 PUT1 GGC1 HIS5 |
| C2 | RPS1B RPL21B ARO2 RPS3 RPS9A RPL18B PSA1 RPL9A RPL13A RPS1A RPL12A RPS16A RPS4B RPS16B YGL149W |
| C3 | ADK1 MDS3 LYS14 |
| C4 | HIS4 ARG1 ODC2 HIS3 YMR321C HOM2 ARO3 ARO4 TMT1 ECM40 TRP3 CPA2 YGL117W |
| C5 | YDR261W-B YDR261C-D YGR038C-B YDR034C-D YPR137C-B YFL002W-A YOR192C-B YDR210C-D YNL054W-B YJR027W |
| C6 | YGL157W GRE2 MCH5 ALT1 YDR056C MET3 |
| C7 | PRE6 CHA1 MAG1 ERO1 PBA1 |
| C8 | PUP3 PDR5 PDR3 SNQ2 REH1 PUT2 RPT6 CDC48 RPT2 SCL1 RPT4 SGT2 RPN9 GET3 |
| C9 | RSB1 PDR12 PDR15 YOR1 YAP1 RPT3 |
| C10 | HCH1 PDR16 HSP10 AHC2 |
| C11 | PDR1 |
| C12 | ARG4 ARG3 ARG5,6 TRP2 ARG8 |
| C13 | ATM1 |
| C14 | PDR10 |
| C15 | PDR11 VPS55 |
| C16 | SSZ1 |
| C17 | PDR17 |
| C18 | RPN10 DDI1 RAD52 |
| C19 | AHP1 UBC4 |
| C20 | STE6 |
| C21 | YCR061W TPO1 YLR326W YCR062W |
| C22 | YMR102C |
| C23 | YPR158C-D YGR035C YER138W-A YDR316W-B YHR214C-B YPR158W-B |
| C24 | RPN12 ICT1 SHP1 CDC53 |
| C25 | MAL32 PGA3 YLR152C MAL12 RAP1 PYC2 |
| C26 | ICY1 STR3 |
| C27 | SCS7 |
| C28 | PCL5 UGA3 |
| C29 | YAR066W |
| C30 | YFL015C |
| C31 | YDR261C-C YER159C-A |
| C32 | RPL22B |
| C33 | VPS61 |
| C34 | YGR293C |
| C35 | YLF2 |
| C36 | RPN13 SBA1 GDS1 UBP6 ILV3 |
| C37 | YGR027W-A |
| C38 | PUS1 NOG1 ECM1 PSP2 PUS7 NMD3 NUG1 LTV1 |
| C39 | ADD66 |
| C40 | ARP4 |
| C41 | YBL101W-B YDR098C-B YBR012W-B YGR161C-D YDR210W-D YGR161W-B YML045W YOR142W-B YDR210W-B YDR365W-B |
| C42 | YMR050C YMR045C YGR027W-B YBL005W-B YML039W |
| C43 | DCW1 |
| C44 | YTH1 |
| C45 | PRE1 PRE8 RPN8 YNL155W PUP1 PRE4 RPN2 |
| C46 | PCI8 |
| C47 | YPL162C |
| C48 | IMD1 RGD1 |
| C49 | YDR210W-C YGR161C-C YFL002W-B YBL005W-A YBL101W-A YDR365W-A YOR343C-B |
| C50 | MAL33 YFR024C |
| C51 | YMR027W |
| C52 | YOL159C-A SER1 |
| C53 | MES1 MRS6 |
| C54 | UIP5 |
| C55 | YNL179C |
| C56 | YFL065C |
| C57 | TUB4 |
| C58 | MAE1 |
| C59 | YLR227W-A YAR010C YPR158W-A YOL103W-A YDR098C-A YML045W-A YLR256W-A YHR214C-C YMR051C YDR316W-A YPR158C-C YER137C-A YBR012W-A YJR026W |
| C60 | CUP2 |
| C61 | MRL1 |
| C62 | BMH1 |
| C63 | TRK2 |
| C64 | DEG1 |
| C65 | YGR137W LSB1 |
| C66 | NRG1 MCT1 |
| C67 | KAR1 |
| C68 | SLF1 TPO4 |
| C69 | SSA2 SSA1 |
| C70 | RPN6 NPL4 PRE9 PRE10 PRE5 |
| C71 | YLR400W YCL019W YBL107W-A YMR158C-B |
| C72 | YLR241W |
| C73 | ELP2 |
| C74 | ERG9 |
| C75 | YDL057W |
| C76 | TEL1 TRP5 |
| C77 | PDI1 MGR1 |
| C78 | YKL069W |
| C79 | BDF1 |
| C80 | JLP2 |
| C81 | YGL204C |
| C82 | PCM1 YBR300C YAT2 |
| C83 | YOR006C YBL028C NOB1 |
| C84 | LSG1 |
| C85 | JIP5 |
| C86 | PAN3 |
| C87 | PLP1 |
| C88 | TRS120 |
| C89 | YGL010W PTC1 |
| C90 | SPE4 |
| C91 | YDR034W-B |
| C92 | YGR164W |
| C93 | RDH54 |
| C94 | YOR060C |
| C95 | ENT4 |
| C96 | KCS1 |
| C97 | YMR046C |
| C98 | ECM21 CMK2 |
| C99 | YJR028W |
| C100 | BUD14 |
| C101 | ELM1 |
| C102 | YDR541C TIR4 |
| C103 | GPI18 |
| C104 | YKU80 |
| C105 | OTU1 |
| C106 | NPA3 CLB3 |
| C107 | FMO1 |
| C108 | SEC1 |
| C109 | ENT1 |
| C110 | GAL83 |
| C111 | YGR111W |
| C112 | HIS1 YBR028C DBP2 YER156C ORT1 GRX4 HOM3 |
| C113 | BLM10 |
| C114 | MET13 ARO8 |
| C115 | OPT1 |
| C116 | YHL029C |
| C117 | ORM2 |
| C118 | MVP1 |
| C119 | LST4 |
| C120 | ADE5,7 |
| C121 | DPH5 |
| C122 | ITC1 |
| C123 | LPD1 |
| C124 | DIA1 |
| C125 | BUG1 |
| C126 | TRM9 |
| C127 | NMD2 |
| C128 | HSV2 |
| C129 | YOR343W-A |
| C130 | UBC13 YPL009C |
| C131 | YOR052C DSS4 |
| C132 | YNR070W |
| C133 | SCJ1 |
| C134 | ECM31 |
| C135 | VHR1 UBX4 |
| C136 | NDE1 |
| C137 | CSN12 |
| C138 | GIS2 SYC1 |
| C139 | YLR225C |
| C140 | SWI6 |
| C141 | TEF2 |
| C142 | YER010C |
| C143 | NIT1 YPL264C YIL165C YHR162W |
| C144 | DSE1 |
| C145 | PSF2 |
| C146 | YJL220W |
| C147 | TEF1 |
| C148 | TDH1 |
| C149 | BIO2 FET3 |
| C150 | ILV1 VAS1 |
| C151 | TDH2 TDH3 |
| C152 | UBP3 |
| C153 | AAP1 |
| C154 | AST1 TRP4 |
| C155 | RIB5 |
| C156 | MER1 |
| C157 | BUD9 |
| C158 | YLR049C |
| C159 | MPD2 RNA14 |
| C160 | YHI9 |
| C161 | SWI1 |
| C162 | YBR147W ADH5 |
| C163 | SKP1 |
| C164 | TYW3 |
| C165 | STR2 ARO1 CPR2 YBR116C |
| C166 | YMC1 |
| C167 | YDR154C |
| C168 | ANT1 |
| C169 | GCR1 |
DDS modeling
We estimated a DDS model using the HMF and the concentrations of representative genes. The DDS model underlying the GRN is an optimal solution after searching all possible directed graphs with 170 nodes and the maximum number of incoming edges (including a possible one from the HMF node) for a gene variable is at most 3. The HMF node is not allowed to have incoming edges. This DDS model captured temporal dependencies among the 169 gene clusters and HMF during the earlier exposure to the inhibitor in yeast fermentation process. A GRN is derived from the DDS model, by creating an edge from each potential regulator j to each gene i if the coefficient of gene j is non-zero in the difference equation of i. The reconstructed GRN of transcriptional regulation with the 169 gene clusters nodes plus an HMF node is partially depicted in Fig. 2. Existence of an edge from Pdr1 to Rib5 indicates a temporal dependency of the rate of change in Rib5 expression on the mRNA level of Pdr1. The number 8 × 10−4, positioned next to the edge, is the p-value of this temporal dependency. The original system matrix was stabilized by scaling all eigenvalues by the spectral norm 2.40. The overall p-value, 0.011, of the fitted DDS model indicates that the model is statistically significant, meaning that the resulting model has high levels of consistency with biological observations because the probability of the model arising by chance is as low as 0.011.
Temporal interactions (partial) of 169 gene clusters in response to HMF treatment for biomass conversion to ethanol by ethanologenic yeast. The p-values of each detected pair of interaction are displayed next to the corresponding edge. A solid directed edge in green from the first gene node to the second gene node with an arrowhead indicates enhancement of the second gene by the first gene; An edge in red from the first gene node to the second gene node with a solid dot indicates repression of the second gene by the first gene. The dashed edges represent the external influence from HMF to each gene: red for repressing and green for enhancing. The graph is rendered by the software GraphViz (www.graphviz.org).
Among the 364 genes in 169 clusters, there are 12 known transcription factors (TFs) according to YEASTRACT [], including Pdr1(C11[1]), Mal33(C50[2]), Cup2 (C60[1]), Nrg1 (C66[2]), Gis2 (C138[2]), Swi6 (C140[1]), Gcr1 (C169[1]), Yap1 (C9[6]), Uga3 (C28[2]), Rap1 (C25[6]), Pdr3 (C8[14]), and Lys14 (C3[3]). We inspect in our DDS model whether these TFs have been identified as of significant temporal influence over other genes as well as whether they show response to HMF treatment. Table 3 lists the number of edges that come out of each TF, as #Detected from the GRN in Fig. 2. It also gives how many among these edges are established transcriptional regulations in yeast in the literature, as #Documented, and the number of potential transcriptional regulations based on sequence motifs, as #Potential. The last column in Table 3 indicates whether a TF has shown statistically significant response to HMF (p-value less than 0.05 for the coefficient of HMF) in the DDS model. Although Table 3, based on the unadjusted p-values of the best fitting difference equations of each gene variable, has not taken into consideration of multiple comparison effects when searching for a best set of influencing nodes, it is useful in eliminating TFs that may not play a significant role in response to HMF. The obvious includes Nrg1 (C66[2]) and Swi6 – No other gene clusters have shown any temporal dependency on either, though Nrg1 may be affected by HMF directly. In addition, a TF that does not directly respond to HMF but does have influences on other genes may less likely participate in the first effect of HMF; these include Cup2, Gcr1, and Mal33.
Table 3
| Known TFs (cluster[#members]) | #Detected | #Documented | #Potential | Subject to HMF Significantly? |
|---|---|---|---|---|
| Cup2 (C60[1]) | 16 | 0 | 2 | No |
| Gcr1 (C169[1]) | 1 | 0 | 1 | No |
| Gis2 (C138[2]) | 3 | 0 | 0 | Yes, negatively |
| Lys14 (C3[3]) | 9 | 0 | 0 | Yes, negatively |
| Mal33 (C50[2]) | 1 | 0 | 0 | No |
| Nrg1 (C66[2]) | 0 | 0 | 0 | Yes, positively |
| Pdr1 (C11[1]) | 16 | 1 | 0 | Yes, positively |
| Pdr3 (C8[14]) | 26 | 0 | 2 | Yes, positively |
| Rap1 (C25[6]) | 1 | 0 | 0 | Yes, negatively |
| Swi6 (C140[1]) | 0 | 0 | 0 | No |
| Uga3 (C28[2]) | 2 | 1 | 0 | Yes, negatively |
| Yap1 (C9[6]) | 8 | 6 | 1 | Yes, positively |
Eight TFs in Table 3 can thus be candidates for responding directly to HMF. However, the p-value must be adjusted to cancel the multiple testing effect in order to reduce the false positive rate. We further used the conservative Bonferroni correction, by inflating all p-values and excluding those interactions with inflated p-values greater than 0.05, to illustrate the most significant interactions in the DDS model (Fig. 3). Two TFs survived the stringent p-value inflation – Pdr3 (C8[14]) and Yap1 (C9[6]). Cluster C8(14), to which Pdr3 belongs, significantly responds to HMF and influences 10 other genes in four clusters. Among those influenced, Top4 (C68[2]) has some motif pattern to which Pdr3p binds, though nothing in literature has been established for the other 9 genes. Cluster C36(5) takes a significant multivariate effect from HMF: HMF has a direct positive role in the gene expression rate of this cluster as well as of cluster C8(14), though C8(14) has a negative effect on C36(5). Thus HMF influences C36(5) in two competing paths and the overall effect is a balance of the two. Cluster C9(6), to which Yap1 belongs, significantly influences a single cluster C45(7). Among the seven genes in C45, six have been experimentally determined as being regulated by Yap1p – Pre1 [], Pre4 [], Pre8 [], Rpn2 [], Rpn8 [], and YNL155w []. Interestingly, four of these six transcriptional interactions regulated by Yap1p in Saccharomyces cerevisiae have been established very recently [] to be highly responsive to the toxicant arsenite at both gene expression and protein levels. Such coincident transcriptional interactions as identified in this study may suggest that Yap1p be involved in the stress tolerance mechanism, and Yap1p can be a core regulator for stress tolerance in yeast. Although Yap1 does not show a significant direct incoming edge from HMF in this new work, its significant interactions downstream found by this study stand still. It suggests a significant role and involvement of Yap1 in response to HMF. In addition, it is encouraging that the GRN model developed in this study is highly consistent with the current knowledge including documented experimental observations and sequence motif based analysis. The other five TFs listed in Table 3, but not appearing in , may still be potentially interesting given the significant correlation with known TFs. However, additional study and supporting data are needed for more conclusive remarks.
Significant temporal interactions of candidate gene clusters in response to HMF treatment for ethanol production by ethanologenic yeast. The adjusted p-values of each edge are displayed. The color scheme follows the previous picture.
In addition to the genes that are directly influenced by HMF, our DDS model also presented numerous interesting interactions among genes with potential significance. For example, cluster C1(5) (Zrt2, Sam3, Put1, Ggc1, His5) showed highly significant negative response to HMF as well as a strong negative influence on the expression rate of ARP4 (). Cluster C106(2) and C26(2) showed a significant negative influence on Ssz1 and cluster C114(2), respectively. Cluster C2(15) has a significant positive influence on the genes in cluster C70(5).
These genes have been observed to be core stress response genes and many related genes are observed to be interesting in coping with the HMF stress for survival [Liu et al., unpublished data]. Resolution of such interactions could have a significant impact to understand the mechanism of detoxification and the stress tolerance caused by HMF. Although they have not been reported, such statistically significant gene interactions presented by this model could be potentially biologically significant to predict unknown gene interactions. With the high consistency of predicted Yap1 and Pdr3 clusters obtained using DDS modeling presented in this study and current knowledge, it is reasonable to assume that relationships predicted using this model are potentially biologically significant. A commonly documented TF Pdr1 shows a possible regulatory role to the selected subset genes in this model. Although it is highly homologous with Pdr3, Pdr1 does not always respond the same with Pdr3. Further examination using biological experiments is needed.
Beyond agreement with existing knowledge of transcriptional regulations in yeast, the interactions discovered in the DDS model are consistent quantitatively with the observed dynamical behavior of our experimental data. Figure 4 shows the DDS model response to HMF and how well the model fits the observed trajectory data for the four clusters that have known transcriptional interactions. Clusters C8(14), C68(2), C9(6), and C45(7) show the prediction of response to HMF with and without HMF (Fig. 4). These clusters all showed strong enhancement in the expression level. The 2nd and 3rd columns in Fig. 4 demonstrate the prediction made by the model and how the time courses evolve differently when the same sample is subject to different experimental conditions. In each plot in the 2nd and 3rd columns, the original time course sample, the log-time interpolated data, and the fitted time course are illustrated. The model captured the trend in the data precisely for all the clusters, given the large sample variation present in most microarray experiments. Visualization of DDS modeling on all other clusters is provided in the .
Simulation of four gene clusters C8(14), C68(2), C9(6), and C45(7) using the reconstructed DDS model. The 1st column displays the predictions of mRNA expression time courses of each cluster without HMF (green solid lines) versus with HMF (red dashed lines). The 2nd column shows the fitting to samples exposed to HMF: Fitted gene expression time courses (red dashed lines) from the model versus the observed ones (yellow dash-dotted lines); the big open triangles or stars represent the original values; the small filled yellow triangles are interpolated values used for model estimation. The 3rd column shows fitting to control samples not exposed to HMF: Fitted gene expression time courses (green solid lines) from the model versus the observed ones (blue dotted lines); the big crosses represent the original values; the small ones are interpolated values actually used.
We also computed a DDS model with at most two incoming edges per gene with an insignificant overall p-value of 0.062. On the other hand, we used a maximum of 4 and 5 incoming edges, more than 3, per gene to derive additional DDS models with denser connectivity. The p-values of each gene variable in the resulting DDS models either decrease very slowly or start to increase as more incoming edges are allowed. We were thus not able to identify any significant interactions after the Bonferroni p-value adjustment. Therefore, we believe that the current complexity of DDS fits the resolution of the data set, and the model has revealed interesting interactions which are worthwhile to undergo further biological validation.
5 Conclusion and future work
We have developed a data-driven DDS modeling framework, by combining concepts in dynamical systems, Markovian chains, multiple linear regression, and combinatorial and least squares optimization, to detect regulatory interactions and to predict system dynamical behaviors based on large-scale data sets. The way that we use the statistical significance, i.e., the p-value, to determine combinatorially the parent assignment of each gene, and the way we stabilize a DDS model have not been seen in the literature to our knowledge. Our modeling strategy can work with non-uniform time-course data, identify a most statistically significant DDS model that is naturally stable when no stimulus is present. Using our DDS modeling in application of yeast transcriptome profiling data challenged by inhibitor HMF, we identified several significant regulatory interactions, among which, transcription factor Yap1 and Pdr3 were significant regulatory elements for HMF tolerance in yeast. Such information aids explanation of yeast adaptation to inhibitors combining with other phenotypes observed previously [, ]. We will apply DDS modeling to recently developed more tolerant strains [] [Liu et al., unpublished] to identify novel interactions for inhibitor tolerance in yeast. Knowledge obtained can guide genetic engineering for stress tolerance strain development. Our DDS modeling methodology can be further applied to analysis of systems from data sets that contain both transcriptome and proteome measured simultaneously on the same sample. Therefore, complete snapshots of molecular processing events can be obtained to provide a more accurate account of the genomic mechanism on inhibitor detoxification and tolerance for ethanologenic yeast.
Supplementary Material
Online Supplemental Materials
Acknowledgments
The project was supported by the National Research Initiative of the USDA Cooperative State Research, Education and Extension Service, grant number 2006-35504-17359. The project described was supported in part by Grant Number 5U54CA132383 to MS from the National Cancer Institute and an Interdisciplinary Research Grant to MS from New Mexico State University. ZO has also been supported in part by the National Science Foundation CREST grant number HRD-0420407. We acknowledge the use of supercomputers IBM BlueGene/L “cyBlue” through Information Infrastructure Institute at Iowa State University and SGI Altix 8200 “Encanto” at New Mexico Computing Applications Center.
Contributor Information
Mingzhou (Joe) Song, Department of Computer Science, New Mexico State University, P. O. Box 30001, MSC CS, Las Cruces, New Mexico 88003, U.S.A.
Zhengyu Ouyang, Department of Computer Science, New Mexico State University, P. O. Box 30001, MSC CS, Las Cruces, New Mexico 88003, U.S.A.
Z. Lewis Liu, National Center for Agricultural Utilization Research, U.S. Department of Agriculture, Agriculture Research Service, 1815 N University Street, Peoria, Illinois 61604, U.S.A.
References
Resourse:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4160033/
Key:Discrete Dynamical System Modeling for Gene Regulatory Networks of HMF Tolerance for Ethanologenic Yeast